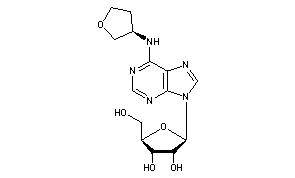
Dasotraline, SEP-225289, DSP-225289
1R,4S Transnorsertraline
Generic Name:Dasotraline
Synonym: SEP-225289
Chemical Name:(1R,4S)-4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1-amine
Synonym: SEP-225289
Chemical Name:(1R,4S)-4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1-amine
4(S)-(3,4-Dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1(R)-ylamine hydrochloride
CAS Number:675126-05-3, Cas of THE DRUG SUBSTANCE hydrochloride is 675126-08-6
Indication:Attention deficit hyperactivity disorder (ADHD)
Drug Company:Sunovion Pharmaceuticals. Inc. in phase 2 as on sept 2014, Sunovion Pharmaceuticals Inc.
CAS Number:675126-05-3, Cas of THE DRUG SUBSTANCE hydrochloride is 675126-08-6
Indication:Attention deficit hyperactivity disorder (ADHD)
Drug Company:Sunovion Pharmaceuticals. Inc. in phase 2 as on sept 2014, Sunovion Pharmaceuticals Inc.
PRONUNCIATION da soe tra’ leen
THERAPEUTIC CLAIM Treatment of attention deficit hyperactivity
disorder (ADHD)
CHEMICAL NAMES
1. 1-Naphthalenamine, 4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydro-, (1R,4S)-
2. (1R,4S)-4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1-amine
THERAPEUTIC CLAIM Treatment of attention deficit hyperactivity
disorder (ADHD)
CHEMICAL NAMES
1. 1-Naphthalenamine, 4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydro-, (1R,4S)-
2. (1R,4S)-4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1-amine
MOLECULAR FORMULA C16H15Cl2N
MOLECULAR WEIGHT 292.2
MOLECULAR WEIGHT 292.2
SPONSOR Sunovion Pharmaceuticals. Inc.
CODE DESIGNATION SEP-225289
CAS REGISTRY NUMBER 675126-05-3
UNII 4D28EY0L5T
WHO NUMBER 9885
CODE DESIGNATION SEP-225289
CAS REGISTRY NUMBER 675126-05-3
UNII 4D28EY0L5T
WHO NUMBER 9885
SEP-225289 is an antidepressant which had been in early clinical trials at Sepracor (now Sunovion Pharmaceuticals) for the treatment of major depressive disorder (MDD). In 2010, the company discontinued development of the compound for this indication. At present, phase II clinical trials are under way for the treatment of attention deficit/hyperactivity disorder (ADHD). In preclinical studies, the drug has been shown to be a potent and balanced reuptake inhibitor of serotonin, norepinephrine and dopamine (SNDRI). A drug candidate with a triple mechanism of action as such may provide a broader spectrum of therapy than currently marketed antidepressants.
Recently, drug candidates for blocking the monoamine reuptake transporters have sparked considerable interest in the pharmaceutical industry for treatment of central nervous system disorders. Various candidates are in clinical evaluation in addition to numerous others at the preclinical stage. Sertraline 2is a selective serotonin reuptake inhibitor (SSRI), marketed by Pfizer as Zoloft for depression. (1R,4S)-4-(3,4-Dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1-amine hydrochloride 1 is structurally similar to sertraline 2 and is currently under investigation for a number of potential central nervous system disorder indications at Sepracor.

ABOUT SERTRALINE

 | |
| (1S,4S)-4-(3,4-dichlorophenyl)-N-methyl-1,2,3,4-tetrahydronaphthalen-1-amine |
 SERTRALINE
SERTRALINE
Clinicians recognize a distinction among central nervous system illnesses, and there have been many schemes for categorizing mental disorders. The Diagnostic and Statistical Manual of Mental Disorders, Fourth Ed., Text Revision, (hereinafter, the “DSM-IV-TR™”), published by the American Psychiatric Association, and incorporated herein by reference, provides a standard diagnostic system upon which persons of skill rely. According to the framework of the DSM-IV-TR™, the CΝS disorders of Axis I include: disorders diagnosed in childhood (such as, for example, attention deficit disorder or “ADD” and attention deficit / hyperactivity disorder or “ADHD”) and disorders diagnosed in adulthood. CΝS disorders diagnosed in adulthood include
(1) schizophrenia and psychotic disorders; (2) cognitive disorders;(3) mood disorders; (4) anxiety related disorders; (5) eating disorders; (6) substance related disorders; (7) personality disorders; and (8) “disorders not yet included” in the scheme.
Of particular interest to the present invention are adulthood disorders of DSM-IN-TR™ categories (1) through (7) and sexual disorders, currently classified in category (8). Mood disorders of particular interest include depression, seasonal affective disorder and bipolar disorder. Anxiety related disorders of particular interest are agoraphobia, generalized anxiety disorder, phobic anxiety, obsessive compulsive disorder (OCD), panic disorder, acute stress disorder, posttraumatic stress disorder, premenstrual syndrome, social phobia, chronic fatigue disorder, perimenopause, menopause and male menopause.
In general, treatment for psychoses, such as schizophrenia, for example, is quite different than treatment for mood disorders. While psychoses are treated with D2 antagonists such as olanzapine (the “typical” and “atypical” antipsychotics), mood disorders are treated with drugs that inhibit the neuronal reuptake of monoamines, in particular, serotonin (5-HT), norepinephrine (ΝE) and dopamine (DA).
[005] Common therapeutic agents for mood disorders include, but are not limited to, selective serotonin reuptake inhibitors (SSRI’s), including fluoxetine, citalopram, nefazodone, fluvoxamine, paroxetine, and sertraline.
Sertraline, whose chemical name (lS,4S)-c/5 4-(3,4-dichlorophenyl)- 1,2,3,4-tetrahydro-Ν-methyl-l-napthalenamine, is approved for the treatment of depression by the United States Food and Drug Administration, and is available under the trade name ZOLOFT® (Pfizer Inc., NY, NY, USA). In the human subject, sertraline has been shown to be metabolized to (lS,4S)-c« 4- (3,4-dichlorophenyl)-l,2,3,4-tetrahydro-l-napthalenamine, also known as desmethylsertraline or norsertraline. Desmethylsertraline has been described as “not contributing significantly to the serotonergic action of sertraline” Ronfield et al, Clinical Pharmacokinetcs, 32:22-30 (1997). Reports from Hamelin et al, Clinical Pharmacology & Therapeutics, 60:512 (1996) and Serebruany et al, Pharmacological Research, 43:453-461 (2001), have stated that norsertraline is “neurologically inactive”. These statements appear to be based on results observed in serotonin-induced syndrome and ptosis in mouse models in vivo, whereas the original Pfizer research papers suggested on the basis of data in vitro that desmethylsertraline was a selective serotonin uptake inhibitor. Koe et al, JPET, 226:686-700 (1983). Sanchez et al, Cellular and Molecular Neurobiology, 19: 467 (1999), speculated that despite its lower potency, desmethylsertraline might play a role in the therapeutic effects of sertraline but, there is presently no evidence in the literature to support this theory.
] The primary clinical use of sertraline is in the treatment of depression. In addition, United States Patent 4,981,870 discloses and claims the use of sertraline and norsertraline, as well as (lR,4S)-trans 4-(3,4-dichlorophenyl)- 1,2,3,4-tetrahydro-N-methyl-l-napthalenamine and (lS,4R)-trαra 4-(3 ,4- dichlorophenyl)- 1 ,2,3 ,4-tetrahydro-N-methyl- 1 -napthalenamine for the treatment of psychoses, psoriasis, rheumatoid arthritis and inflammation. The receptor pharmacology of the individual (1S,4R) and (1R,4S) enantiomers of trα«5 4-(3,4-dichlorophenyl)-l,2,3,4-tetrahydro-N-methyl-l -napthalenamine is described by Welch et al, J. Med. Chem., 27:1508-1515 (1984). Summary of the Invention
It has now been discovered that {\R,4S)-trans 4-(3,4-dichlorophenyl)- 1,2,3,4-tetrahydro-l-napthalenamine (P) and (lS,4R)-tra«_ 4-(3 ,4- dichlorophenyl)- 1,2,3, 4-tetrahydro-l-napthalenamine (Q) are useful in the treatment of CNS-related disorders that are modulated by monoamine activity, and produce diminished side effects as compared to the current standards of treatment. Treatable CNS disorders include, but are not limited to, mood disorders {e.g., depression), anxiety disorders {e.g., OCD), behavioral disorders {e.g., ADD and ADHD), eating disorders, substance abuse disorders and sexual function disorders. The compounds are also useful for the prophylaxis of migraine.
Compounds P and Q are represented by the formulae:

In one aspect, the present invention relates to a method for treating CNS disorders, which involves the administration of a therapeutically effective amount of P or Q, or a pharmaceutically acceptable salt of either.
In another aspect, the invention relates to trans- 4-(3,4-dichlorophenyl)- 1,2,3,4-tetrahydro-l-napthalenamine of the formula (PQ):
NH2

(PQ)

Skeletal formulae of chlorprothixene and tametraline, from which sertraline was derived

Norsertraline, sertraline’s chief active metabolite
………………………………………..
PATENT
(Scheme 2).

In a preferred embodiment, the compound prepared by the route of Scheme 2 is (1R,4S)-trans 4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydro-1-naphthalenamine. Even more preferred is the preparation of the compound substantially free of its cis isomer.
Example 1
Synthesis of N—((S)-4-(3,4-dichlorophenyl)-3,4-dihydronaphthalen-1-yl)acetamide (3)1.1. Synthesis of Oxime 2
A suspension formed from a mixture of (S)-tetralone 1 (56.0 g, 0.192 mol), hydroxylamine hydrochloride (14.7 g, 0.212 mol), and sodium acetate (17.4 g, 0.212 mol) in methanol (168 mL) was heated to reflux for 1 to 5 hours under a N2atmosphere. The progress of the reaction was monitored by HPLC. After the reaction was complete, the reaction mixture was concentrated in vacuo. The residue was diluted with toluene (400 mL) and 200 mL water. The organic layer was separated and washed with an additional 200 mL water. The organic layer was concentrated and dried to give crude solid oxime 2 (58.9 g, 100%), m. p. 117-120° C.
1H NMR (400 MHz, CDCl3) δ (ppm) 9.17 (br, 1H, OH), 7.98 (m, 1H), 7.36 (d, 1H, J=8.0 Hz), 7.29 (m, 2H), 7.20 (d, 1H, J=2.4 Hz), 6.91 (m, 2H), 4.11 (dd, 1H, J=7.2 Hz, 4.4 Hz), 2.82 (m, 2H), 2.21 (m, 1H), 2.08 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 154.94, 144.41, 140.40, 132.83, 130.92, 130.82, 130.68, 130.64, 129.98, 129.38, 128.12, 127.64, 124.48, 44.52, 29.51, 21.27.
1.2. Synthesis of Enamide 3
The solution of the crude oxime 2 (59 g, 0.193 mol) in toluene (500 mL) was purged with N2 for 30 min. Et3P (25 g, 0.212 mol) was charged. After stirring for 10 min, acetic anhydride (21.6 g, 20 mL, 0.212 mol) was added. The reaction mixture was refluxed for 8 to 13 h. Progress of the reaction was monitored by HPLC. The reaction mixture was cooled to room temperature. 6N NaOH (aq) (86 mL, 0.516 mol) and 1.0 M (n-Bu)4NOH in methanol (1.0 mL) were added. The hydrolysis was complete in about 2 to 4 h. The organic layer was separated and diluted with EtOAc (300 mL) and 2-BuOH (30 mL). The diluted organic solution was washed with 1% HOAc (aq) solution (300 mL) and DI water (3×300 mL) and concentrated to about 350 mL of a slurry in vacuo. The slurry was diluted with heptane (100 mL) and 2-BuOH (4 mL) and heated to reflux to form a clear solution. Heptane (50 to 200 mL) was slowly added until a cloudy solution formed. The suspension was slowly cooled to rt. The product was filtered out, washed with 30% toluene and 70% heptane (3×100 mL) solution and dried in a vacuum oven to give 56.9 g white solid (enamide 3, 89% yield), m. p. 167-168° C.
(S)-Tetralone 1 (50.0 g, 0.172 mol) was slurried in methanol (150 mL) with hydroxylamine hydrochloride (13.1 g, 0.189 mol) and sodium acetate (15.5 g, 0.189 mol). The resulting suspension was heated to reflux for 2 to 6 h under an inert atmosphere with progress monitored by HPLC. On completion, the mixture was cooled to 25° C., diluted with toluene (300 mL) and quenched with 1.7 N NaOH (100 mL). The mixture was concentrated in vacuo under reduced pressure, the aqueous layer removed and the organic layer washed further with DI water (100 mL). Further toluene (300 mL) was charged to the vessel and water removed by azeotropic distillation. Once at ambient temperature, n-Bu3P (47.1 mL, 0.183 mol) was charged to the reactor, followed by acetic anhydride (32.5 mL, 0.344 mol). The reaction was heated to reflux and monitored by HPLC. After 20-24 h, the reaction was cooled to ambient temperature and quenched with 6 N NaOH (120 mL). This mixture was allowed to react for 2 to 6 h before the aqueous layer was removed. The organic phase was washed with DI water (100 mL). Concentration of the mixture in vacuo, cooling to room temperature and diluting with isopropanol (50 mL) was done prior to addition of heptane to assist with crystallization. An initial charge of heptane (50 mL) was followed by an additional 650 mL. Aging of the slurry followed by filtration, washing (4×100 mL heptane) and drying yielded a light yellow solid (enamide 3, 44.1 g, 77%).
1H NMR (400 MHz, CDCl3) δ (ppm) 7.35 (d, 1H, J=8.4 Hz), 7.26 (m, 3H), 7.17 (m, 1H), 7.05 (dd, 1H, J=8.0, 1.6 Hz), 7.00 (br, 1H), 6.87 (m, 0.82H, 82% NH rotamer), 6.80 (br, 0.18H, 18% NH rotamer), 6.31 (t, 0.82H, J=4.8 Hz, 82% H rotamer), 5.91 (br, 0.18H, 18% H rotamer), 4.12 (br, 0.18H, 18% H rotamer), 4.03 (t, 0.82H, J=8.0 Hz, 82% H rotamer), 2.72 (m, 1H), 2.61 (ddd, 1H, J=16.8, 8.0, 4.8 Hz), 2.17 (s, 2.46H, 82% CH3 rotamer), 1.95 (s, 0.54H, 18% CH3rotamer). 100 MHz13CNMR (CDCl3) δ 169.3, 143.8, 137.7, 132.3, 131.8, 131.4, 130.5, 130.3, 130.2, 128.8, 128.1, 127.8, 127.2, 123.8, 122.5, 121.2, 117.5, 42.6, 30.3, 24.1.
Example 2Synthesis of N-((1R,4S)-4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1-yl)acetamide (4)
The enamide 3 (24 g, 72 mmol) was slurried in degassed isopropanol (200 mL). The resulting slurry was transferred to the appropriate reactor. Prior to the addition of the catalyst solution, the content of the reactor was purged with nitrogen. A solution of (R,R)-MeBPE(COD)RhBF4 catalyst (20.1 mg, 0.036 mmol, 0.05 mol %) in isopropanol (IPA) (100 mL) was added to the reactor. The content was cooled to 0° C. and purged with nitrogen three times. The reactor was then purged with hydrogen and pressurized to 90 psig. The reaction was aged with agitation at 0° C. for 7.5 h and conversion was monitored by the hydrogen uptake. The content was then warmed to RT and hydrogen was vented. After purging with nitrogen, the contents were drained. The reaction mixture was heated to 50° C. and filtered through a pad of Celite. The clear orange solution was concentrated to ˜50% volume (150 mL) and diluted with toluene (5.9 g, 5 wt %). The suspension was heated to 65° C. and water (14.7 mL) was added dropwise to form a cloudy solution. The slurry was slowly cooled to −10° C. and aged for 30 minutes. The solid was filtered and washed with cold IPA (2×45 mL). The cake was dried under vacuum at 45° C. overnight to afford 20.0 g (83% yield) of trans acetamide 4 (>99% de).
1H NMR (CDCl3) 400 MHz δ 7.34 (dd, 2H, J=7.9, 2.4 Hz), 7.23 (t, 1H, J=7.5 Hz), 7.15 (m, 2H), 6.85 (dd, 1H, J=8.2, 2.0 Hz), 6.82 (d, 1H, J=7.7 Hz), 5.72 (d, 1H, J=8.4 Hz), 5.31 (dd, 1H, J=13.2, 8.1 Hz), 4.10 (dd, 1H, J=7.0, 5.9 Hz), 2.17 (m, 2H), 2.06 (s, 3H), 1.87 (m, 1H). 1.72 (m, 1H); 13C NMR (CDCl3) 100 MHz δ 169.7, 146.9, 138.8, 137.7, 132.6, 130.8, 130.6, 130.5, 130.3, 128.4, 128.3, 127.9, 127.4, 47.9, 44.9, 30.5, 28.4, 23.8.
Example 3
Synthesis of (1R,4S)-4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1-amine Hydrochloride (5)
A solution of trans-acetamide 4 (9.0 g, 26.9 mmol), n-propanol (45 mL) and 5M hydrochloric acid (45 mL) was refluxed for approximately 48 h (90-93° C.). During this time, the reaction temperature was maintained at ≧90° C. by periodically collecting the distillate until the reaction temperature was >92° C. Additional n-propanol was added periodically to maintain the solution at its original volume. After the hydrolysis was complete, the solution was slowly cooled to 0° C., resulting in a slurry, which was aged for one hour at 0° C. The reaction mixture was filtered, and the cake was washed with 1:1 methanol/water (20 mL), followed by t-butyl methyl ether (20 mL). The wet-cake was dried under vacuum at 45 to 50° C. to afford 7.0 g of the amine hydrochloride 5 (80% yield).
1H NMR (DMSO-d6) δ 1.81-1.93 (m, 2H), 2.12-2.21 (m, 1H), 2.28-2.36 (m, 1H), 4.28 (t, 1H, J=6.8), 4.59 (br.s, 1H), 6.84 (d, 1H, J=7.6), 7.05 (dd, 1H, J=8.4, 1.6), 7.25 (t, 1H, J=7.6), 7.32 (t, 1H, J=7.6), 7.37 (d, 1H, J=1.6), 7.56 (d, 1H, J=8.4), 7.76 (d, 1H, J=7.2), 8.80 (br.s, 3H);
13C NMR (DMSO-d6) 147.4, 138.9, 133.6, 131.0, 130.5, 130.4, 130.1, 129.0, 128.9, 128.4, 128.2, 126.8, 47.9, 43.1, 27.8, 25.2.
INTERMEDIATE
Example 5 Catalytic Asymmetric Hydrogenation of the Enamide 3 Using (R,S,R,S)-MePenn Phos(COD)RhBF4 as the Catalyst
As shown in Scheme 4, the enamide 3 was subjected to homogeneous catalytic asymmetric hydrogenation in the presence of a chiral catalyst, H2, and a solvent. In this example the catalyst was derived from the complex of the transition metal rhodium with the chiral phosphine ligand, (1R,2S,4R,5S)—P,P-1,2-phenylenebis {(2,5-endo-dimethyl)-7-phosphabicyclo[2.2.1]heptane}(R,S,R,S-MePennPhos). The hydrogenations were carried out at a substrate concentration of about 0.12 M to about 0.24 M of compound 3.

………………………………………..
Koenig, Stefan G.; Vandenbossche, Charles P.; Zhao, Hang; Mousaw, Patrick; Singh, Surendra P.; Bakale, Roger P.
Organic Letters, 2009 , vol. 11, 2 pG . 433 – 436
Organic Letters, 2009 , vol. 11, 2 pG . 433 – 436

Imidoyl chlorides, generated from secondary acetamides and oxalyl chloride, can be harnessed for a selective and practical deprotection sequence. Treatment of these intermediates with 2 equiv of propylene glycol and warming enables the rapid release of amine hydrochloride salts in good yields. Notably, the reaction conditions are mild enough to allow for a swift deprotection with no observed epimerization of the amino center.
Supporting Information A Facile Deprotection of Secondary Acetamides
(1R,4S)-4-(3,4-Dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1-amine hydrochloride – Compound 1, Scheme 1 / Table 3, entry 1A:
decomp. > 290 °C.
1H NMR (400 MHz, DMSO-d6) δ 8.71 (s, 3H), 7.71 (d, 1H, J = 7.7 Hz), 7.53 (d, 1H, J = 8.1 Hz), 7.34 (s, 1H),
7.29 (m, 1H), 7.22 (m, 1H), 7.01 (d, 1H, J = 8.1 Hz), 6.81 (d, 1H, J = 7.7 Hz), 4.56 (s,
1H), 4.26 (s, 1H), 2.26 (m, 1H), 2.15 (m, 1H), 1.83 (m, 2H).
7.29 (m, 1H), 7.22 (m, 1H), 7.01 (d, 1H, J = 8.1 Hz), 6.81 (d, 1H, J = 7.7 Hz), 4.56 (s,
1H), 4.26 (s, 1H), 2.26 (m, 1H), 2.15 (m, 1H), 1.83 (m, 2H).
13C NMR (100 MHz, DMSO-d6) δ 147.3, 138.8, 133.5, 130.9, 130.5, 130.4, 130.0, 128.9, 128.8, 128.3, 128.1,
126.7, 47.8, 43.0, 27.7, 25.1.
126.7, 47.8, 43.0, 27.7, 25.1.
NMR GRAPHS GIVEN
13 C NMR

………………………………………..
Jerussi, T. P.; Fang, Q. K.; Currie, M. G. PCT Int. Appl. WO 2004042669 A1 200440325, 2004.

The discovery route involved preparation of (S)-tetralone (4S)-3 from racemic tetralone(4RS)-3 via chromatographic separation of sulfinyl imine (Rs,4RS)-5diastereomers, followed by hydrolysis. The sulfinyl imine isomers were generated by condensation with (R)-tert-butylsulfinamide ((R)-TBSA), (Rs)-4, in the presence of titanium ethoxide. The yield of sulfinyl imine diastereomer (Rs,4S)-5 was ∼15% after chromatographic purification. The low recovery yield was due to chromatographic loss and the instability of compound 5 on silica gel. The resulting (S)-tetralone (4S)-3 was converted to N-formyl amine (1RS,4S)-6 as a mixture of two diastereomers that were again separated by chromatography to afford the desired diastereomer(1R,4S)-6 in 17% yield over two steps. (1R,4S)-trans-norsertraline 1 was obtained after the acidic hydrolysis of (1R,4S)-6 in 71% yield. The overall yield of this route was less than 2% and involved two chromatographic purifications, making it impractical for an efficient large-scale synthesis of 1.
Jerussi, T. P.; Fang, Q. K.; Currie, M. G. PCT Int. Appl. WO 2004042669 A1 200440325, 2004.http://www.google.com/patents/WO2004024669A1?cl=en
……………………………………………
PAPER
Development of a large-scale stereoselective process for (1R,4S)-4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1-amine hydrochloride
Org Process Res Dev 2007, 11(4): 726
Org Process Res Dev 2007, 11(4): 726

A convenient, multikilogram-scale, stereoselective process for the synthesis of (1R,4S)-4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1-amine hydrochloride 1 is described. The key steps involve synthesis of sulfinyl imine(Rs,4S)-5 from (S)-tetralone (4S)-3 and (R)-tert-butylsulfinamide (Rs)-4, and its stereoselective reduction with 9-BBN to produce the (1R)-amine center of 1. The process has been scaled up to multikilogram scale and gives 1 in an overall yield of >50% with a chemical purity of 99.7 A% by HPLC and stereochemical purity of >99.9% by chiral HPLC.
(1R,4S)-4-(3,4-Dichlorophenyl)-1,2,3,4-tetrahydronaphthalen-1-ylamine HCl (1).
1H NMR (400 MHz, DMSO-d6) δ 1.81−1.93 (m, 2H), 2.12−2.21 (m, 1H), 2.28−2.36 (m, 1H), 4.28 (t, 1H, J = 6.8 Hz), 4.59 (br s, 1H), 6.84 (d, 1H, J = 7.6 Hz), 7.05 (dd, 1H, J = 8.4, 1.6 Hz), 7.25 (t, 1H, J = 7.6 Hz), 7.32 (t, 1H, J = 7.6 Hz), 7.37 (d, 1H, J = 1.6 Hz), 7.56 (d, 1H, J = 8.4 Hz), 7.76 (d, 1H, J = 7.2 Hz), 8.80 (br s, 3H).
13C NMR (100 MHz, DMSO-d6) δ 147.4, 138.9, 133.6, 131.0, 130.5, 130.4, 130.1, 129.0, 128.9, 128.4, 128.2, 126.8, 47.9, 43.1, 27.8, 25.2.
Anal. Calcd for C16H15Cl2N: C, 58.47; H, 4.91; N, 4.26; Cl, 32.36. Found: C, 58.44; H, 4.79; N, 4.21; Cl, 32.53.
……………………………………………………………..
WO 2004024669
Preparation of compounds of the present invention is illustrated below in Scheme 1 and its accompanying narrative.

[0015] In the compound

of Scheme 1,
R is “R,° , wherein R1, R2 and R3 are each independently alkyl. In a preferred embodiment of the compounds, R is tert-butyl.
[0016] N-[4-(3 ,4-dichlorophenyl)- 1 ,2,3 ,4-tefrahydronaphthalen- 1 -yl]formamide, the intermediate in the synthesis shown in Scheme 1 , exists in four stereoisomeric forms:

C (1S,4S) D (1R.4R) [0017] When N-[4-(3 ,4-dichlorophenyl)- 1 ,2,3,4-tetrahydronaρhthalen-l - yl]formamide is synthesized from achiral starting materials via non- stereoselective syntheses, all four isomers will be produced. The mixture can be readily separated into a racemic cis diastereomer and a racemic trans diastereomer by means, such as recrystallization or chromatography on achiral media, that rely on chemical and physical differences.
[0018] The trans diastereomer, represented as E below, is a 1 :1 mixture of A and B. When E is hydrolyzed, PQ is produced; when A is hydrolyzed, P is produced; when B is hydrolyzed, Q is produced. The cis diastereomer, represented as F below, is a 1 : 1 mix of C and D.

E = A + B F = C + D
……………………………………………………………………………………………
WO 2007006003

Scheme 3
Production (lR,4S)-4-(3,4-dichloro-phenyl)-l,2,3,4-tetrahydro-naphthalen-l-ylamine HCl from 4-(S)-(3,4-dichloro-phenyl)-3,4-dihydro-2H-naphthalen-l-one.

(S)-(3,4-Dichloro-phenyl)-3,4- (1 R,4S)-4-(3,4-Dichloro-phenyl)-1 ,2,3,4-tetrahydro- d ιhydro-2H-naphthalen-1 -one naphthalen-1 -ylamine; [0080] Charge 4-(S)-(3,4-dichloro-phenyl)-3,4-dihydro-2H-naρhthalen-l-one (1 kg, 3.4 mol) and (R)-tert-butylsulphinamide (TBSA, 464 g, 3.8 mol) to a suitable reactor and dissolved in about 7 L THF. Add a 20%wt solution of Titanium ethoxide in ethanol (about 7.8 kg, 6.9 mol) and heat the mixture to about 70 0C for about 24h. The reaction is monitored by HPLC, and after the reaction is complete, cool the mixture to room temperature and added a 24% wt aqueous solution of NaCl to the mixture. The resultant slurry was filtered and washed multiple times with about 1 L total of ethyl acetate. The mother liquors and washes were concentrated to a minimum volume. The aqueous phase was extracted with about 5 L of ethyl acetate and evaporated to dryness.
[0081] The contents were then dissolved in about 7 L of THF and cooled to about —10 0C. About 9 kg, (~5 mol) of a 0.5 M solution of 9-borabicycIononane (9-BBN) in THF, was added slowly (about 3h) and the mixture was stirred at 00C until reaction completion. A 6N HCl/methanol (~2L) was added to the mixture and stirred until the hydrolysis reaction was complete. After neutralization with about 2 L of 6N aqueous NaOH, the mixture was distilled to remove THF and the residue (aqueous phase) was extracted twice with methyl t- butyl ether (2x6L). The organic phase was then washed with water. The organic phase was concentrated, then cooled to 00C followed by addition of 2N HCl in methyl t-butyl ether (3 L). The product slowly precipitated as the HCl salt during the addition. The slurry was filtered and washed with methyl t-butyl ether (2x2L). The product was dried under vacuum at about 45°C to afford about 850 g of Re-Crystallization of crude (lR,4S)-4-(3,4-dichloro-phenyl)- 1,2,3,4-tetrahydro-naphthalen-l-ylamine HCl.
[0082] The resulting (lR,4S)-4-(3,4-dichloro-phenyl)-l,2,3,4-tetrahydro- naphthalen-1-ylamine HCl (85Og) was charged to a suitable reactor and about 30 L of denatured ethanol was added. The mixture was heated to reflux, the volume was reduced to about 50% via distillation, and then cooled to 50°C. About 30 L of Hexane was added to the slurry to complete the product crystallization and then the slurry was cooled to about 00C. The product was isolated by filtration, the cake was washed with about 2 L of ethanol/hexane (1/3 v/v) and then about 2 L of ethyl acetate, followed by about 3 L of hexane. The wet cake was dried under vacuum at about 45°C to afford 630 g of product.
[0083] Another alternative process for preparation of compound P is presented below.
[0084] 4-(S)-(3,4-dichloro-phenyl)-3,4-dichloro-2H-naphthalen-l-one (4.11 kg) and (R)-tert-butylsulphinamide (TBSA, 1.9 kg) were charged to a suitable reactor and dissolved in 29 L THF. A 20%wt solution of titanium ethoxide in ethanol (31.6 kg) was added and the mixture was heated to 70 °C with stirring. The reaction is monitored by HPLC, and after the reaction was complete (20-24 h) the mixture was cooled to room temperature and added to 20 L of a 24 wt% aqueous solution of NaCl. The resultant slurry was filtered and washed 3 times with ethyl acetate (4.1 L). The mother liquors and washes were concentrated to a minimum volume. The aqueous phase was extracted with about 20 L of a 1 :1 mix of ethyl acetate and toluene. The organic phases were combined and concentrated to half volume to give a solution of 2. A purified sample of 2 was analyzed: m.p. 104 0C, 1HNMR (400 MHz, CDCl3) δ (ppm) 8.23 (dd, IH, J= 7.9, 0.9 Hz), 7.38 (ddd, IH, J= 14.7, 7.3, 1.5 Hz), 7.37 (d, IH, J= 8.4 Hz), 7.33 (d, IH, J= 7.7 Hz), 7.17 (d, IH, J= 1.8 Hz), 6.93 (d, IH, J= 7.7 Hz), 6.89 (dd, IH, J= 8.4, 2.2 Hz), 4.18 (dd, IH, J= 7.3, 4.8 Hz), 3.36 (ddd, IH, J= 17.5, 8.8, 4.4 Hz), 2.93 (ddd, IH, J= 17.6, 8.3, 4.2 Hz), 2.33 (m, IH), 2.15 (m, IH), 1.34 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 175.8, 144.2, 142.7, 132.6, 130.8, 130.7, 129.7, 128.1, 127.6, 127.4, 57.8, 44.3, 31.1, 29.4, 22.8. HRMS calc for C20H2ICl2NOS 394.0799, found 394.0767.
[0085] The solution of imine (2) was cooled to -10 0C and 36.3 kg of a 0.5 M solution of 9-borabicyclononane (9-BBN) in THF, was added slowly (over 3h) and the mixture was stirred at 0 0C until reaction completion. A 4N HCl/methanol (8 L) was added to the mixture and stirred until the hydrolysis reaction was complete. After neutralization with about 15 kg of 6N aqueous NaOH (pH 8), the mixture was distilled to remove THF and methanol. The residue (aqueous phase) was extracted twice with methyl t-butyl ether (2 x 16L). The organic phase was then washed with water. The organic phase was concentrated, then cooled to 00C followed by addition of 2N HCl in methyl t- butyl ether (5.4 kg). The product precipitated as the HCl salt. The slurry was filtered, washed with methyl t-butyl ether (2 x 8L) and dried under vacuum at 450C to afford about 3.73 kg of crude (lR,4S)-4-(3,4-dichloro-phenyl)-l,2,3,4- tetrahydro-naphthalen-1-ylamine HCl (compound P).
A purified sample of P was analyzed: NOTE P IS DASOTRALINE
m.p. 152 – 154 0C,
1H NMR (400 MHz, CDCl3) δ (ppm) 7.58 (d, IH, J= 7.7 Hz), 7.29 (m, 2H), 7.18 (br. t, IH, J= 7.5 Hz), 7.09 (d, IH, J= 1.8 Hz), 6.87 (d, IH, J= 7.7 Hz), 6.80 (dd, IH, J= 8.3, 2.0 Hz), 4.65 (dd, IH, J= 4.4, 4.4 Hz), 4.15 (t, IH, J= 5.5 Hz), 3.30 (d, IH, J= 3.7 Hz), 2.35 (m, IH), 1.95 (m, IH), 1.85 (m, IH), 1.75 (m, IH), 1.23 (s, 9H).
13C NMR (100 MHz, CDCl3) δ 147.1, 138.4, 138.0, 132.6, 130.8, 130.6, 130.5, 129.8, 128.3, 127.9, 55.8, 53.3, 44.0, 28.2, 27.7, 22.9.
HRMS calc for C20H23Cl2NOS 396.0956, found 396.0968.
[0086] The crude (lR,4S)-4-(3,4-dichloro-phenyl)-l,2,3,4-tetrahydro- naphthalen-1-ylamine HCl (3.63 kg) was charged to a suitable reactor and 128 L of denatured ethanol was added. The mixture was stirred at reflux and polish filtered. The volume was reduced to about 50% via distillation, and then cooled to 500C. 80 L of heptane was added to the slurry to complete the product crystallization and then the slurry was cooled to -5 °C. The product was filtered, the cake was washed twice with 5.7 L of ethanol/heptane (1/1 v/v) and then washed with 6 L of hexane. The wet cake was dried under vacuum at about 45°C to afford 2.57 kg of product. The product had a chemical purity of 99.65 A% and a diastereomeric purity in excess of 99%
…………………………………………………………………………
PATENT
WO 2011069032
Transnorsertraline, i. e. , (1 R,4S)-trans-4-(3 ,4-dichlorophenyl)- 1 ,2,3 ,4-tetrahydro- 1 – naphthalenamine and (lS,4R)-trans-4-(3,4-dichlorophenyl)-l,2,3,4-tetrahydro-l- naphthalenamine are described in, for example, U.S. Patent No. 7,087,785 B2 (“the ‘785 patent”; incorporated herein by reference in its entirety), have the following chemical structures, respectively:

Uses of transnorsertraline in the treatment, prevention, or management of affective disorders and other various CNS disorders are also disclosed in the ‘785 patent. Such disorders include, but are not limited to, depression, mood disorders, anxiety disorders, behavioral disorders, eating disorders, substance abuse disorders, and sexual function disorders.
ref
A Randomized, Double-Blind, Parallel-Group, Multicenter Efficacy and Safety Study of SEP-225289 Versus Placebo in Adults With Attention Deficit Hyperactivity Disorder (ADHD) (NCT01692782)
ClinicalTrials.gov Web Site 2012, September 27
ClinicalTrials.gov Web Site 2012, September 27
Characterization of the electrophysiological properties of triple reuptake inhibitors on monoaminergic neurons
Int J Neuropsychopharmacol 2011, 14(2): 211
Int J Neuropsychopharmacol 2011, 14(2): 211
PET evaluation of serotonin and dopamine transporter occupancy associated with administration of SEP-225289
Biol Psychiatry 2010, 67(9, Suppl. 1): Abst 102
[65th Annu Meet Soc Biol Psychiatry (SOBP) (May 20-22, New Orleans) 2010]
Biol Psychiatry 2010, 67(9, Suppl. 1): Abst 102
[65th Annu Meet Soc Biol Psychiatry (SOBP) (May 20-22, New Orleans) 2010]
Koenig, Stefan G.; Vandenbossche, Charles P.; Zhao, Hang; Mousaw, Patrick; Singh, Surendra P.; Bakale, Roger P.
Organic Letters, 2009 , vol. 11, (2) pg 433 – 436
Organic Letters, 2009 , vol. 11, (2) pg 433 – 436
Thalen, Lisa K.; Zhao, Dongbo; Sortais, Jean-Baptiste; Paetzold, Jens; Hoben, Christine; Baeckvall, Jan-E.
Chemistry – A European Journal, 2009 , vol. 15, ( 14) pg. 3403 – 3410
Chemistry – A European Journal, 2009 , vol. 15, ( 14) pg. 3403 – 3410
| US8134029 | Jul 30, 2010 | Mar 13, 2012 | Sunovion Pharmaceuticals Inc. | Treatment of CNS disorders with trans 4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydro-1-napthalenamine |
| US8658700 | Dec 4, 2012 | Feb 25, 2014 | Sunovion Pharmaceuticals Inc. | Treatment of CNS disorders with trans 4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydro-1-napthalenamine |
| US20010044474* | Dec 20, 2000 | Nov 22, 2001 | Curatolo William J. | Hydrogel-driven layered drug dosage form |
| US20060257475* | Aug 17, 2006 | Nov 16, 2006 | Boehringer Ingelheim International Gmbh | Stable Sertraline Hydrochloride Formulation and Method |
| US20080280993* | Jul 15, 2008 | Nov 13, 2008 | Sepracor Inc. | Treatment of CNS Disorders With trans 4-(3,4-Dichlorophenyl)-1,2,3,4-Tetrahydro-1-Napthalenamine |